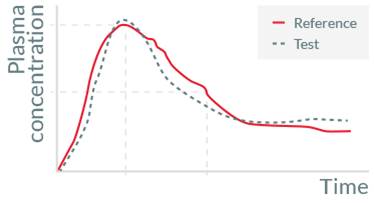
生物等效性实验 (BE):是通过对比仿制制剂和对照制剂 (原研药) 在人体内吸收的程度和速度的异同,来判定仿制制剂与原研制剂的药效学异同,体内一致可以说是仿制药临床前研究阶段的终极目标。
但由于生物等效性实验 (BE) 试验的复杂性决定了试验的进行时间周期长、费用高,因此在研发实际的操作过程中,对每一批次的样品都进行生物等效性实验 (BE) 测试显然是不现实的,特别在处方和工艺摸索阶段中更为明显。
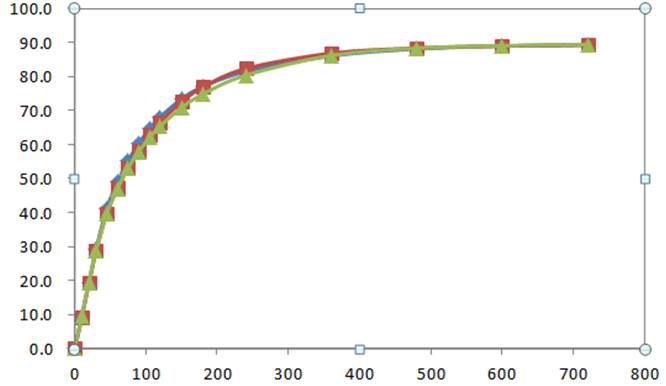
而体外的溶出度试验,可以有效地反映体内的溶出情况,因而是药品在处方和工艺筛选阶段,以及上市产品质量控制阶段的一个有效方式。